Research of the Kirchner Group
Overview
- Cluster weighting and development of software PEACEMAKER1
- Development of software TRAVIS2
- (Vibrational) Spectroscopy in complex systems and condensed phases3
- Working principles of ionic liquids4
- Molecular level insight into deep eutectic solvents5
- Functional materials: Chemically active domains in liquids6
- Bulk and interfacial liquid-liquid systems for extraction and energy devices7
Research topics constantly change - sometimes slow sometimes fast. Please refer also to our publication page8 in order to obtain an overview over the most recent research topics.


QCE and Peacemaker
If a molecule possesses many conformers and we are interested in how much a particular conformer contributes to a certain property we want to address weights to the molecules. This is usually done by Boltzmann weighting which applies for the same system size, i.e., different conformers of one monomer. If we increase the system by including different oligomers, e.g. compare the contribution of a dimer with that of a monomer, we are unable to apply Boltzmann factors. Instead we can use cluster weighting. As in Boltzmann weighting, models of statistical thermodynamics are combined with quantum chemically calculated molecules – here, clusters of different size or oligomers. With this approach we can describe neat liquids and their mixtures at non-zero temperature and pressure in the condensed and gaseous phase. Upon inclusion of clusters with different composition, e.g. a solute solvated
with different amounts of solvent molecules, concentration dependent properties are accessible as well. Self-consistent-field calculations lead to equilibrium populations of these clusters and thus an ensemble of different structural states is generated similar to molecular dynamics simulations.
List of papers: 2019, 20010, 19911, 19012, 18713, 18414, 17115, 15016, 12717, 11218, 10019, 9920, 7521, 7422, 6523, 3624.
Development of software TRAVIS
Our open source software program TRAVIS (TRajectory Analyzer and VISualizer) offers various functions for the easy and fast investigation of static (structural), dynamic, spectroscopic and miscellaneous system properties.
Paper examples:
- TRAVIS — A free analyzer for trajectories from molecular simulation (215)
TRAVIS (TRajectory Analyzer and VISualizer) – which started as the master project of Dr. Martin Brehm – is a program package for post-processing and analyzing trajectories from molecular dynamics and Monte Carlo simulations. It is an open source free software licensed under the GNU GPL, it is platform independent, and does not require any external libraries. The many functions that are implemented cover standard analysis, both structural and dynamical as well as novel functions as the void or reactive flux analyses. Functions for predicting vibrational spectra from molecular dynamics simulations are also part of TRAVIS (see 3)). People who worked or currently work on TRAVIS during their PhD and others: Dr. Martin Brehm, Dr. Martin Thomas, Sascha Gehrke, and Vahideh Alizadeh.
Keywords: Analysis software, post-processing of trajectories - Robustness of the hydrogen bond and ion pair dynamics in ionic liquids to different parameters from the reactive flux method (212)
Many properties of liquids are originated in the dynamic interplay of molecular interactions as hydrogen bonds or electrostatic forces. However, the quantification of these dynamics is challenging, due to perturbations from other processes, like diffusion. The reactive flux methodology is a sophisticated approach to eliminate this interference.
Recently, we adapted the reactive flux ansatz and developed tools for the analysis of the dynamics of hydrogen bonds and ionic networks from it (see 189), which were implemented in our in-house program package TRAVIS. In this latest publication we present an exhaustive benchmark study to demonstrate the stability of our implementation to several settings. Thereby, the superiority of the method compared to other, easier approaches was impressively demonstrated.
Keywords: Non-covalent interactions, hydrogen bond dynamics, reactive flux, ion pair dynamics
List of papers: 21525, 21226, 20627, 20428, 19529, 18930, 16931, 16632, 16433, 16334, 15635, 14936, 14537, 13938, 13339, 11940, 11441, 11342, 10943, 10844, 9245, 9046, 8147, 8048, 4049.

(Vibrational) Spectroscopy in complex systems and condensed phases
Molecular spectroscopy provides valuable information, e.g. on the structure of molecules, the composition of a given sample, the concentration of substances therein, and the mechanism and rate of chemical reactions. The theoretical prediction of vibrational spectra does not only help interpreting experimental spectra, but based on molecular simulations it additionally provides deeper insight into the molecular vibrations of flexible molecules and their interplay with the environmental medium. Different techniques, like infrared (IR), Raman, vibrational circular dichroism (VCD) can be routinely applied via our analysis tool TRAVIS. While TRAVIS follows the autocorrelation appraoch, we also provide an alternative tool with the quantum cluster equilibrium (QCE) method (Link to our QCE Code Peacemaker). QCE averages over the spectra of different clusters, which can consist of different oligomers and different compositions. This allows the concentration-dependent determination of spectra.
Working Principles of ionic liquids (ILs)
Ionic liquids (ILs) are a fascinating material class, because they are entirely composed of mobile ions and they are salts that possess a wide liquid range at low temperatures. Understanding the manifoldness of interactions governing ILs and their mixtures as well as the resulting properties is crucial for their use in different applications, like material synthesis, electrochemistry, energy devices, extraction processes and many more. Gaining deeper insight into their working principles helps to improve the potential of ILs to serve as better alternative to conventional solvents.
List of papers: 21151, 20352, 19853, 19754, 19655, 19456, 19357, 19158, 18930, 18859, 18660, 18561, 18262, 18163, 17964, 17865, 17666, 17467, 17268, 16869, 16770, 16571, 16433, 16272, 16073, 15974, 15775, 15576, 15477, 15278, 15179, 14880, 14781, 14537, 14482, 14383, 14284, 14185, 13938, 13886, 13787, 13688, 13489, 13290, 12991, 12892, 12693, 12594, 12495, 12296, 11940, 11897, 11598, 11342, 11199, 107100, 106101, 104102, 103103, 95104, 91105, 91105, 9046, 89106, 88107, 85108, 82109, 8048, 79110, 69111, 67112, 66113, 64114, 57115, 55116, 49117, 48118, 44119, 43120, 42121, 41122, 34123, 25124.

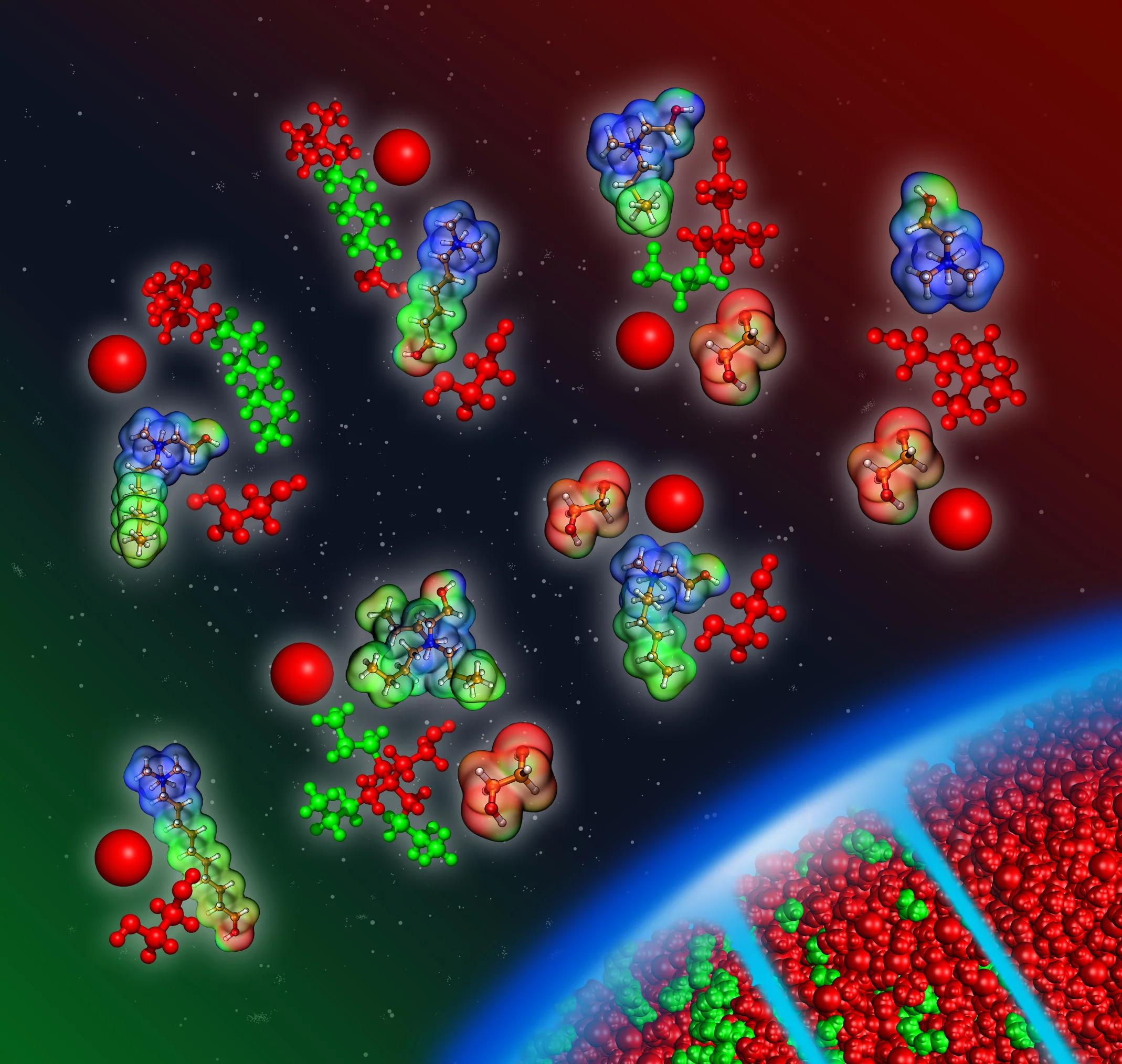
Molecular level insight into deep eutectic solvents (DES)
Deep eutectic solvents (DES) are mixtures of two or more substances which liquefy at a much lower temperature than the primary substances melting points (eutectic point). They are classified according to several main types depending on their constituents. DES are distinguished from usual eutectic mixtures and highlighted with the deep prefix, because their melting temperature is supposed to be significantly lower than the ideal eutectic temperature. Moreover, defining a mixture as a deep eutectic solvent is not simple and the current definitions need to be extended nowadays in order to address the nature of DES more rigorously. DES display the essential advantage of having good environmental bio-compatibility and low toxicity. They are a cheap, sustainable and promising alternatives to ionic liquids and conventional liquids in various processes. Despite the fact that DES as designer solvents possess wide range applications, there is a lack of fundamental information about them.
Functional materials: Chemically active domains in liquids
Ionic liquids or deep eutectic solvents can act as functional materials. Depending on the chemical nature of the liquids’ components, special structural motives emerge on the molecular level, while macroscopically the system remains a homogeneous liquid. This is for example the case if alkyl chain functionalizations at the molecular compounds are introduced. The liquid reveals a segregation into polar and nonpolar domains, which allow for the development of new reaction routes in these solvents.


Bulk and interfacial liquid-liquid and liquid-solid systems for extraction and energy devices
Sustainable processes to counteract environmental problems request the development of neoteric solvents for extraction and recycling processes as well as energy devices like batteries, super capacitors and solar cells. For this purpose it is mandatory to fully understand the complex interactions at liquid-liquid and liquid-solid interfaces. In that regard – if modeled properly – molecular simulations can provide deeper insight and thus help to develop novel solvents and strategies for more sustainable applications and technologies.
List of papers: 210130, 208131, 207128, 19853, 19529, 175132, 16770, 16073, 14383.
Links
- https://www.chemie.uni-bonn.de/kirchner/de/research#QCE
- https://www.chemie.uni-bonn.de/kirchner/de/research#Travis
- https://www.chemie.uni-bonn.de/kirchner/de/research#Vibration
- https://www.chemie.uni-bonn.de/kirchner/de/research#IL
- https://www.chemie.uni-bonn.de/kirchner/de/research#DES
- https://www.chemie.uni-bonn.de/kirchner/de/research#Domain
- https://www.chemie.uni-bonn.de/kirchner/de/research#Energy-device
- https://www.chemie.uni-bonn.de/kirchner/de/publications
- https://www.chemie.uni-bonn.de/kirchner/de/publications#201
- https://www.chemie.uni-bonn.de/kirchner/de/publications#200
- https://www.chemie.uni-bonn.de/kirchner/de/publications#199
- https://www.chemie.uni-bonn.de/kirchner/de/publications#190
- https://www.chemie.uni-bonn.de/kirchner/de/publications#187
- https://www.chemie.uni-bonn.de/kirchner/de/publications#184
- https://www.chemie.uni-bonn.de/kirchner/de/publications#171
- https://www.chemie.uni-bonn.de/kirchner/de/publications#150
- https://www.chemie.uni-bonn.de/kirchner/de/publications#127
- https://www.chemie.uni-bonn.de/kirchner/de/publications#112
- https://www.chemie.uni-bonn.de/kirchner/de/publications#100
- https://www.chemie.uni-bonn.de/kirchner/de/publications#99
- https://www.chemie.uni-bonn.de/kirchner/de/publications#75
- https://www.chemie.uni-bonn.de/kirchner/de/publications#74
- https://www.chemie.uni-bonn.de/kirchner/de/publications#65
- https://www.chemie.uni-bonn.de/kirchner/de/publications#36
- https://www.chemie.uni-bonn.de/kirchner/de/publications#215
- https://www.chemie.uni-bonn.de/kirchner/de/publications#212
- https://www.chemie.uni-bonn.de/kirchner/de/publications#206
- https://www.chemie.uni-bonn.de/kirchner/de/publications#204
- https://www.chemie.uni-bonn.de/kirchner/de/publications#195
- https://www.chemie.uni-bonn.de/kirchner/de/publications#189
- https://www.chemie.uni-bonn.de/kirchner/de/publications#169
- https://www.chemie.uni-bonn.de/kirchner/de/publications#166
- https://www.chemie.uni-bonn.de/kirchner/de/publications#164
- https://www.chemie.uni-bonn.de/kirchner/de/publications#163
- https://www.chemie.uni-bonn.de/kirchner/de/publications#156
- https://www.chemie.uni-bonn.de/kirchner/de/publications#149
- https://www.chemie.uni-bonn.de/kirchner/de/publications#145
- https://www.chemie.uni-bonn.de/kirchner/de/publications#139
- https://www.chemie.uni-bonn.de/kirchner/de/publications#133
- https://www.chemie.uni-bonn.de/kirchner/de/publications#119
- https://www.chemie.uni-bonn.de/kirchner/de/publications#114
- https://www.chemie.uni-bonn.de/kirchner/de/publications#113
- https://www.chemie.uni-bonn.de/kirchner/de/publications#109
- https://www.chemie.uni-bonn.de/kirchner/de/publications#108
- https://www.chemie.uni-bonn.de/kirchner/de/publications#92
- https://www.chemie.uni-bonn.de/kirchner/de/publications#90
- https://www.chemie.uni-bonn.de/kirchner/de/publications#81
- https://www.chemie.uni-bonn.de/kirchner/de/publications#80
- https://www.chemie.uni-bonn.de/kirchner/de/publications#40
- https://www.chemie.uni-bonn.de/kirchner/de/publications#214
- https://www.chemie.uni-bonn.de/kirchner/de/publications#211
- https://www.chemie.uni-bonn.de/kirchner/de/publications#203
- https://www.chemie.uni-bonn.de/kirchner/de/publications#198
- https://www.chemie.uni-bonn.de/kirchner/de/publications#197
- https://www.chemie.uni-bonn.de/kirchner/de/publications#196
- https://www.chemie.uni-bonn.de/kirchner/de/publications#194
- https://www.chemie.uni-bonn.de/kirchner/de/publications#193
- https://www.chemie.uni-bonn.de/kirchner/de/publications#192
- https://www.chemie.uni-bonn.de/kirchner/de/publications#188
- https://www.chemie.uni-bonn.de/kirchner/de/publications#186
- https://www.chemie.uni-bonn.de/kirchner/de/publications#185
- https://www.chemie.uni-bonn.de/kirchner/de/publications#182
- https://www.chemie.uni-bonn.de/kirchner/de/publications#181
- https://www.chemie.uni-bonn.de/kirchner/de/publications#179
- https://www.chemie.uni-bonn.de/kirchner/de/publications#178
- https://www.chemie.uni-bonn.de/kirchner/de/publications#176
- https://www.chemie.uni-bonn.de/kirchner/de/publications#174
- https://www.chemie.uni-bonn.de/kirchner/de/publications#172
- https://www.chemie.uni-bonn.de/kirchner/de/publications#168
- https://www.chemie.uni-bonn.de/kirchner/de/publications#167
- https://www.chemie.uni-bonn.de/kirchner/de/publications#165
- https://www.chemie.uni-bonn.de/kirchner/de/publications#162
- https://www.chemie.uni-bonn.de/kirchner/de/publications#160
- https://www.chemie.uni-bonn.de/kirchner/de/publications#159
- https://www.chemie.uni-bonn.de/kirchner/de/publications#157
- https://www.chemie.uni-bonn.de/kirchner/de/publications#155
- https://www.chemie.uni-bonn.de/kirchner/de/publications#154
- https://www.chemie.uni-bonn.de/kirchner/de/publications#153
- https://www.chemie.uni-bonn.de/kirchner/de/publications#151
- https://www.chemie.uni-bonn.de/kirchner/de/publications#148
- https://www.chemie.uni-bonn.de/kirchner/de/publications#147
- https://www.chemie.uni-bonn.de/kirchner/de/publications#144
- https://www.chemie.uni-bonn.de/kirchner/de/publications#143
- https://www.chemie.uni-bonn.de/kirchner/de/publications#142
- https://www.chemie.uni-bonn.de/kirchner/de/publications#141
- https://www.chemie.uni-bonn.de/kirchner/de/publications#138
- https://www.chemie.uni-bonn.de/kirchner/de/publications#137
- https://www.chemie.uni-bonn.de/kirchner/de/publications#136
- https://www.chemie.uni-bonn.de/kirchner/de/publications#134
- https://www.chemie.uni-bonn.de/kirchner/de/publications#132
- https://www.chemie.uni-bonn.de/kirchner/de/publications#129
- https://www.chemie.uni-bonn.de/kirchner/de/publications#128
- https://www.chemie.uni-bonn.de/kirchner/de/publications#126
- https://www.chemie.uni-bonn.de/kirchner/de/publications#125
- https://www.chemie.uni-bonn.de/kirchner/de/publications#124
- https://www.chemie.uni-bonn.de/kirchner/de/publications#122
- https://www.chemie.uni-bonn.de/kirchner/de/publications#118
- https://www.chemie.uni-bonn.de/kirchner/de/publications#115
- https://www.chemie.uni-bonn.de/kirchner/de/publications#111
- https://www.chemie.uni-bonn.de/kirchner/de/publications#107
- https://www.chemie.uni-bonn.de/kirchner/de/publications#106
- https://www.chemie.uni-bonn.de/kirchner/de/publications#104
- https://www.chemie.uni-bonn.de/kirchner/de/publications#103
- https://www.chemie.uni-bonn.de/kirchner/de/publications#95
- https://www.chemie.uni-bonn.de/kirchner/de/publications#91
- https://www.chemie.uni-bonn.de/kirchner/de/publications#89
- https://www.chemie.uni-bonn.de/kirchner/de/publications#88
- https://www.chemie.uni-bonn.de/kirchner/de/publications#85
- https://www.chemie.uni-bonn.de/kirchner/de/publications#84
- https://www.chemie.uni-bonn.de/kirchner/de/publications#79
- https://www.chemie.uni-bonn.de/kirchner/de/publications#69
- https://www.chemie.uni-bonn.de/kirchner/de/publications#67
- https://www.chemie.uni-bonn.de/kirchner/de/publications#66
- https://www.chemie.uni-bonn.de/kirchner/de/publications#64
- https://www.chemie.uni-bonn.de/kirchner/de/publications#57
- https://www.chemie.uni-bonn.de/kirchner/de/publications#55
- https://www.chemie.uni-bonn.de/kirchner/de/publications#49
- https://www.chemie.uni-bonn.de/kirchner/de/publications#48
- https://www.chemie.uni-bonn.de/kirchner/de/publications#44
- https://www.chemie.uni-bonn.de/kirchner/de/publications#43
- https://www.chemie.uni-bonn.de/kirchner/de/publications#42
- https://www.chemie.uni-bonn.de/kirchner/de/publications#41
- https://www.chemie.uni-bonn.de/kirchner/de/publications#34
- https://www.chemie.uni-bonn.de/kirchner/de/publications#25
- https://www.chemie.uni-bonn.de/kirchner/de/publications#209
- https://www.chemie.uni-bonn.de/kirchner/de/publications#183
- https://www.chemie.uni-bonn.de/kirchner/de/publications#177
- https://www.chemie.uni-bonn.de/kirchner/de/publications#207
- https://www.chemie.uni-bonn.de/kirchner/de/publications#180
- https://www.chemie.uni-bonn.de/kirchner/de/publications#210
- https://www.chemie.uni-bonn.de/kirchner/de/publications#208
- https://www.chemie.uni-bonn.de/kirchner/de/publications#175